

EMA dopuszcza do obrotu szczepionki Pfizer i Moderna
Dotychczasowe warunkowe zezwolenie zamieniono na standardowe - firmy nie muszą już wnioskować co roku o odnowienie zezwolenia.

16 września - bez zbytniego nagłośnienia w mediach - pojawiła się na stronie Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych informacja Prezesa Urzędu „w sprawie rekomendacji Europejskiej Agencji Leków (EMA) dotyczącej przekształcenia dotychczasowych warunków pozwoleń na dopuszczenie do obrotu szczepionek Comirnaty i Spikevax w pozwolenia standardowe”.

Oznacza to, że warunkowe dotychczas pozwolenie na dopuszczenie do obrotu (conditional marketing authorisation) szczepionek przeciw COVID-19 Comirnaty (szczepionka firmy BioNTech/Pfizer) i Spikevax (szczepionka firmy Moderna) zostaje przekształcone w standardowe pozwolenie i producenci szczepionek nie będą musieli każdego roku starać się o odnowienie zezwolenia.
Europejska Agencja Leków (EMA) wyjaśnia na swoich stronach, że ponieważ wspiera rozwój leków, które odpowiadają na niezaspokojone potrzeby medyczne, to w interesie zdrowia publicznego wnioskodawcy mogą otrzymać warunkowe pozwolenie na dopuszczenie do obrotu leków na podstawie mniej kompleksowych danych klinicznych niż normalnie wymagane, w przypadku gdy korzyść z natychmiastowej dostępności leku przeważa nad ryzykiem związanym z faktem, że nadal wymagane są dodatkowe dane.
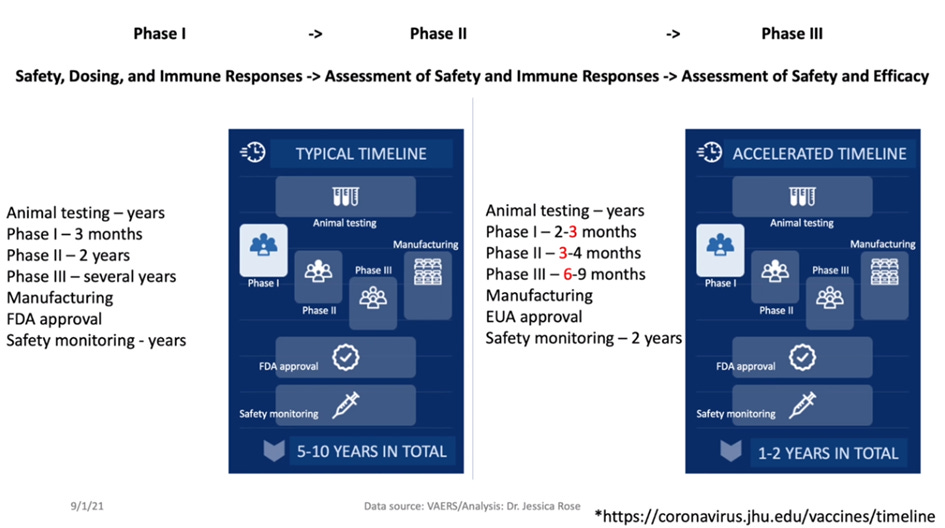
Typowy czas opracowywania szczepionki wynosi od 5 do 10 lat (może nawet trwać dłużej). Jest to konieczne, aby właściwie ocenić bezpieczeństwo i skuteczność danego leku biologicznego w badaniach klinicznych, potrzebny jest także czas na zatwierdzenie leku przez organy regulacyjne, trzeba wyprodukować odpowiednią ilość produktu, aby móc go dystrybuować. Niezwykle ważne jest oszacowanie kwestii bezpieczeństwa, skuteczności i ryzyka, które jest miarą prawdopodobieństwa wystąpienia zdarzenia niepożądanego lub jakiegoś niepożądanego wyniku oraz wagi wynikającej z tego szkody dla zdrowia jednostki w populacji. Bezpieczeństwo jest zatem oceną dopuszczalności tego ryzyka w określonej sytuacji, np. przy wprowadzaniu na rynek nowego produktu biologicznego. Tak więc typowy czas od badań na zwierzętach do monitorowania bezpieczeństwa wynosi około 5-10 lat. Badania na zwierzętach mogą trwać lata, mogą też trwać miesiące - zależy to od tego, jak rozwijają się dane laboratoryjne. Badania I fazy, w których bierze udział niewielka liczba osób, trwają zazwyczaj kilka miesięcy, a po ich zakończeniu - jeśli w ogóle je przeprowadzono - przechodzi się do badań II fazy, w których bierze udział większa liczba osób. Jeśli jednak badania II fazy przebiegną prawidłowo i nie wystąpi zbyt wiele zdarzeń niepożądanych, można przejść do badań III fazy, w których bierze udział znacznie większa grupa osób. Trwa to zwykle kilka lat. Po zakończeniu badań III fazy, jeśli zakończą się one sukcesem, przechodzi się do procesu produkcji i zatwierdzania, a te dwa etapy są ze sobą ściśle powiązane. Następnie produkty są monitorowane przez wiele lat za pomocą narzędzi nadzoru nad bezpieczeństwem farmakoterapii, takich jak VAERS.
Obecnie mamy do czynienia z tak zwaną przyspieszoną linią czasu. Jest to nowa koncepcja, a stworzono ją, ponieważ w marcu 2020 WHO ogłosiła, że świat stoi w obliczu kryzysu i ogłoszono globalną pandemię. Łączny czas trwania badania III fazy prowadzonego przez firmę Pfizer BioNTech wynosił sześć miesięcy (!). Nie były przeprowadzane badania na zwierzętach przed badaniami fazy I, badania fazy II i I były mieszane. Badania fazy II trwały tylko trzy miesiące, co jest to możliwe, ale niedopuszczalne jest mieszanie faz badania. Nie bez powodu są one uporządkowane sekwencyjnie. Zatem łączny czas trwania badania III fazy firmy Pfizer BioNTech wyniósł sześć miesięcy...
Każde badanie kliniczne I, II i III fazy ma listę kryteriów wykluczenia, a listy te są zazwyczaj bardzo długie. Zawsze znajdują się na nich kobiety w ciąży, kobiety karmiące lub kobiety w wieku rozrodczym, ponieważ po prostu nie chcemy zaszkodzić tym osobom. Badanie kliniczne (firmy Pfizer) rzeczywiście uwzględniało na liście kryteriów wykluczenia kobiety w ciąży, kobiety karmiące, osoby z obniżoną odpornością oraz osoby z chorobami autoimmunologicznymi, co oznacza, że nie mogły one uczestniczyć w tych sześciomiesięcznych badaniach. Jest to bardzo ważne, ponieważ definicja bezpieczeństwa została opracowana na podstawie danych z zaledwie sześciu miesięcy. Jak to więc możliwe, że wstrzykuje się te preparaty kobietom w ciąży? Dzieciom? Osobom z chorobami nowotworowymi? Nie mamy żadnych danych dotyczących bezpieczeństwa.
EMA pisze w uzasadnieniu decyzji, że obie szczepionki otrzymały warunkowe pozwolenia w momencie ich dopuszczenia do obrotu, co nałożyło na firmy obowiązki przedstawiania wyników z trwających badań klinicznych oraz dostarczania dodatkowych danych dotyczących jakości farmaceutycznej szczepionek w świetle planowanego zwiększenia skali produkcji. A także, że „Powyższe analizy i dodatkowe badania, w tym badania obserwacyjne, dostarczyły potwierdzające dane dotyczące kluczowych aspektów, przede wszystkim, w szczególności w jak wysokim stopniu szczepionki zapobiegają ciężkiej postaci COVID-19. Ponadto firmy dostarczyły wszystkie wymagane dodatkowe dane dotyczące jakości farmaceutycznej szczepionek”.
EMA dokonuje przeglądu i publikuje – cyklicznie uaktualniany – plik COMIRNATY (COVID-19 mRNA VACCINE) RISK MANAGEMENT PLAN – Plan Zarządzania Ryzykiem - https://www.ema.europa.eu/en/documents/rmp-summary/comirnaty-epar-risk-management-plan_en.pdf, z którego możemy się dowiedzieć, że z badań klinicznych wyłączone były:
- osoby szczepione jakąkolwiek szczepionką przeciwko koronawirusom
- osoby u których stwierdzono wcześniejsze kliniczne lub mikrobiologiczne rozpoznanie COVID-19
- osoby z upośledzoną odpornością, ze znanym lub podejrzewanym niedoborem odporności
- osoby, które przyjmowały produkty krwi/osocza lub immunoglobuliny w okresie 60 dni przed podaniem interwencji w ramach badania interwencyjnym lub planowanym przyjmowaniem w czasie trwania badania
- kobiety w ciąży lub karmiące piersią (powód wykluczenia: aby uniknąć stosowania w populacji wrażliwej).
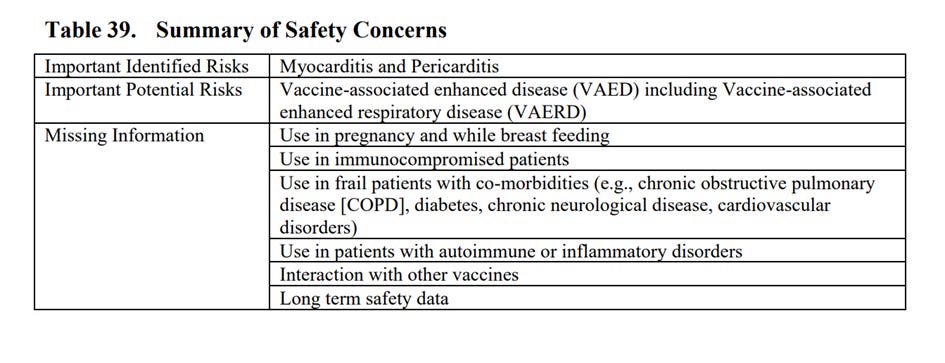
W dokumencie wyszczególnione są (od str. 85) także szczegóły dotyczące ważnych zidentyfikowanych zagrożeń, ważnych potencjalnych zagrożeń i brakujących informacji. I tak ważnym zidentyfikowanym ryzykiem jest zapalenie mięśnia sercowego i osierdzia (końcowy raport z badania klinicznego przewidziany na 31 marca 2026 r.), potencjalnym ważnym ryzykiem wzmożona choroba związana ze szczepionką (Vaccine-Associated Enhanced Disease - VAED),
w tym rozszerzona choroba układu oddechowego związana ze szczepionką (Vaccine-Associated Enhanced Respiratory Disease - VAERD) – końcowy raport z badania przewidziany na 31 grudnia 2023 r.
Brakujące informacje, które Pfizer wymienia w dokumencie to stosowanie w ciąży i podczas karmienia piersią, stosowanie u pacjentów z obniżoną odpornością, stosowanie u pacjentów osłabionych, ze współistniejącymi chorobami (np. przewlekłą obturacyjną chorobą płuc, cukrzycą, przewlekłą chorobą neurologiczną, zaburzeniami sercowo-naczyniowymi), stosowanie u pacjentów z zaburzeniami autoimmunologicznymi lub zapalnymi, interakcje z innymi szczepieniami oraz dane dotyczące bezpieczeństwa długoterminowego (strony 95 – 96).
W informacji Prezesa Cessaka czytamy: „Biorąc pod uwagę całość dostępnych danych dotyczących skuteczności i bezpieczeństwa wynikających z powszechnego wykorzystania tych szczepionek, szczególne zobowiązania nie są już uważane za kluczowe do oszacowania stosunku korzyści do ryzyka tych produktów, co otworzyło drogę do przejścia z warunkowego na standardowe pozwolenia na dopuszczenie do obrotu. Warunkowe pozwolenia na dopuszczenie do obrotu podlegają corocznemu przeglądowi i przedłużeniu ważności. CHMP zalecił ich zamianę na standardowe pozwolenia w wyniku drugiej corocznej procedury przedłużenia ważności pozwoleń. Zalecenie to obejmuje wszystkie istniejące i mające powstać adaptowane szczepionki Comirnaty i Spikevax, w tym niedawno zatwierdzone adaptowane szczepionki Comirnaty Original/Omicron BA.1, Comirnaty Original/Omicron BA.4/5 i Spikevax biwalentna Original/Omicron BA.1.”
Wynika z tego, że według EMA i Prezesa Cessaka nie trzeba już testować produktów na ludziach, że badania kliniczne prowadzone na 8 do 12 myszach to wystarczająca przesłanka do udzielenia standardowego pozwolenia na użycie (biwalentne szczepionki na wariant omikron BA.4/5, które były testowane na 8 do 12 myszach, a na żadnym człowieku).
Prezes Cessak informuje też nas, że warunkowe pozwolenia na dopuszczenie do obrotu zostały przyznane m.in. w oparciu o prawdopodobieństwo, że wnioskodawcy będą w stanie dostarczyć bardziej kompleksowe dane kliniczne i jakościowe po wydaniu pozwolenia na dopuszczenie do obrotu, co uznano za konieczne, biorąc pod uwagę nowatorski sposób działania szczepionek i ich bardzo duże przewidywane zastosowanie.
Patrząc na coraz to nowe dowody dotyczące fałszowania danych z badań klinicznych (Brook Jackson vs. Ventavia), analizy krytyczne bioststystyków (Christtine Cotton) pokazujące, że wyników ogłoszonych przez farmaceutycznego giganta nie można uznać ani za "wiarygodne" ani za "uczciwe" w odniesieniu do Dobrych Praktyk Klinicznych, coraz częstszą krytykę dotychczasowych zwolenników szczepień (Dr Aseem Malhotra), ujawniane dane dotyczące olbrzymiej skali zdarzeń niepożądanych po szczepieniach (ostatnio ujawnione przez CDC dane z aplikacji V-safe) – trudno oprzeć się wrażeniu, że EMA oraz urząd Pana Cessaka przestały pełnić funkcję regulatora dbającego o dobro pacjentów, a zajęły się działalnością marketingową i sprzedażową w imieniu gigantów farmaceutycznych.
W przypadku EMA to nie powinno dziwić. EMA czyli Europejska Agencja Leków (European Medicines Agency) jest zdecentralizowaną agencją Unii Europejskiej (UE) odpowiedzialną za naukową ocenę, nadzór i monitorowanie bezpieczeństwa leków w UE. EMA jest zarządzana przez niezależny zarząd. Bieżące działania prowadzone są przez personel EMA, pod nadzorem dyrektora wykonawczego EMA. Jest organizacją sieciową, w której działalność zaangażowane eksperci z całej Europy.
Zarząd EMA składa się z 36 członków, powołanych do działania w interesie publicznym, którzy nie reprezentują żadnego rządu, organizacji ani sektora. Kogo zatem reprezentuje w EMA Pan Cessak skoro jako Prezes URPLWMiPB jest “centralnym organem administracji rządowej”? Zarząd ustala budżet Agencji, zatwierdza roczny program pracy i jest odpowiedzialny za zapewnienie efektywnej pracy i udanej współpracy Agencji z organizacjami partnerskimi w całej UE i poza nią. Skoro EMA jest agencją UE to pewnie finansowanie pochodzi ze źródeł unijnych? W 2022r całkowity budżet Europejskiej Agencji Leków (EMA) wynosi 417,5 mln EUR. Tylko 13% z wkładu to pieniądze Unii Europejskiej (UE) przeznaczone na kwestie zdrowia publicznego. Około 86% budżetu Agencji pochodzi z opłat i należności pobieranych za usługi regulacyjne (czyli za opłaty pochodzące od firm farmaceutycznych za wnioski o rejestrację nowych produktów leczniczych), a mniej niż 1% z innych źródeł. Z całego budżetu w 2022 r: około 357,7 mln EUR spodziewane jest z opłat i należności; spodziewane są dochody z UE w wysokości około 55,2 mln EUR, głównie w celu wsparcia polityki w zakresie leków sierocych i pediatrycznych, terapii zaawansowanych oraz mikro-, małych i średnich przedsiębiorstw. Zdecydowana większość wpływów do budżetu EMA pochodzi od firm farmaceutycznych, które składają wnioski o rejestracje nowych leków. Czy istnieje bardziej korupcjogenny układ? W aneksie 2 do raportu finansowego za rok 2018-2019 jest wykaz kwalifikujących się branżowych organizacji interesariuszy (stan na dzień 23 lipca 2020 r.) – wymienionym udziałowcem jest na przykład Vaccine Europe, które zrzesza „największe innowacyjne, oparte na badaniach naukowych firmy produkujące szczepionki, działające w Europie, jak również małe i średnie przedsiębiorstwa, takie jak: Moderna, Novavax, Pfizer, AstraZeneca, Janssen, Sanofi”.
Agencja pobiera opłatę za rozpatrywanie wniosków od przedsiębiorstw, które chcą wprowadzić lek na rynek. Pobiera również opłaty za usługi związane z wprowadzaniem leków do obrotu w UE w takich dziedzinach, jak doradztwo naukowe, kontrole i ustalanie najwyższych dopuszczalnych poziomów pozostałości.
Agencja koordynuje naukową ocenę wniosków i związane z tym prace z krajowymi organami regulacyjnymi ds. leków w państwach członkowskich UE. Agencja wynagradza organy krajowe za tę pracę oraz za udział członków ich personelu w komitetach naukowych Agencji, grupach roboczych i innych działaniach. Szacuje się, że w 2022 r. z budżetu Agencji krajowym agencjom regulacyjnym ds. leków zostanie wypłacone 143,1 mln EUR. Czy to oznacza, że EMA płaci na utrzymanie urzędu, którego prezesem jest Pan Cessak? Czy funkcja członka zarządu EMA jest opłacana przez agencję czy jest to funkcja pełniona społecznie?
Co tak naprawdę oznacza ten ruch – to się okaże. Być może to ostatnia faza pandemicznej farsy, decyzja, która normalizuje stan do tej pory traktowany jako tymczasowe zagrożenie. Jednak tryb budzi wątpliwości o przyszłość – co jeszcze zaaplikuje nam EMA jako panaceum na przyszłe dolegliwości? Kiedy przywrócimy właściwą rolę agencjom regulacyjnym, którą powinna być dbałość o dobro pacjenta, jego bezpieczeństwo i minimalizację ryzyka?
Źródła:
https://christine-cotton.1ere-page.fr/expertise-in-english/
https://www.ema.europa.eu/en/about-us/who-we-are
https://www.ema.europa.eu/en/about-us/how-we-work/governance-documents/funding